top of page

eduo
visual
Cell Biology
Connective Tissue Biochemistry: Collagen Types (I, II, III, IV), Elastin, Proteoglycans, Fibronectin, Laminin — Synthesis Defects (Osteogenesis Imperfecta, Ehlers-Danlos, Marfan, Scurvy)
Core Principle of Connective Tissue Biochemistry
Connective tissue provides structural support through an extracellular matrix composed of fibrous proteins (collagen, elastin) and ground substance (proteoglycans, glycosaminoglycans).
Collagen is the most abundant protein in the body, providing tensile strength; elastin provides elasticity; proteoglycans create a hydrated gel that resists compression.
Cell adhesion molecules like fibronectin and laminin connect cells to the matrix and facilitate signaling.
Defects in these components → characteristic clinical syndromes with predictable patterns based on which tissues rely most heavily on the affected protein.
Board pearl: Understanding normal structure-function relationships predicts clinical manifestations of defects.

Collagen Structure and Assembly
Collagen begins as procollagen with a triple helix of three α chains, each containing the repetitive sequence Gly-X-Y (where X is often proline, Y is often hydroxyproline or hydroxylysine).
Glycine at every third position is essential — its small size allows tight helix packing. Any substitution → structural instability.
Post-translational modifications occur in the ER/Golgi: hydroxylation of proline and lysine (requires vitamin C), glycosylation of hydroxylysine.
Procollagen is secreted, terminal propeptides are cleaved → tropocollagen, which self-assembles into fibrils.
Cross-linking via lysyl oxidase (requires copper) creates mature, insoluble collagen fibers.

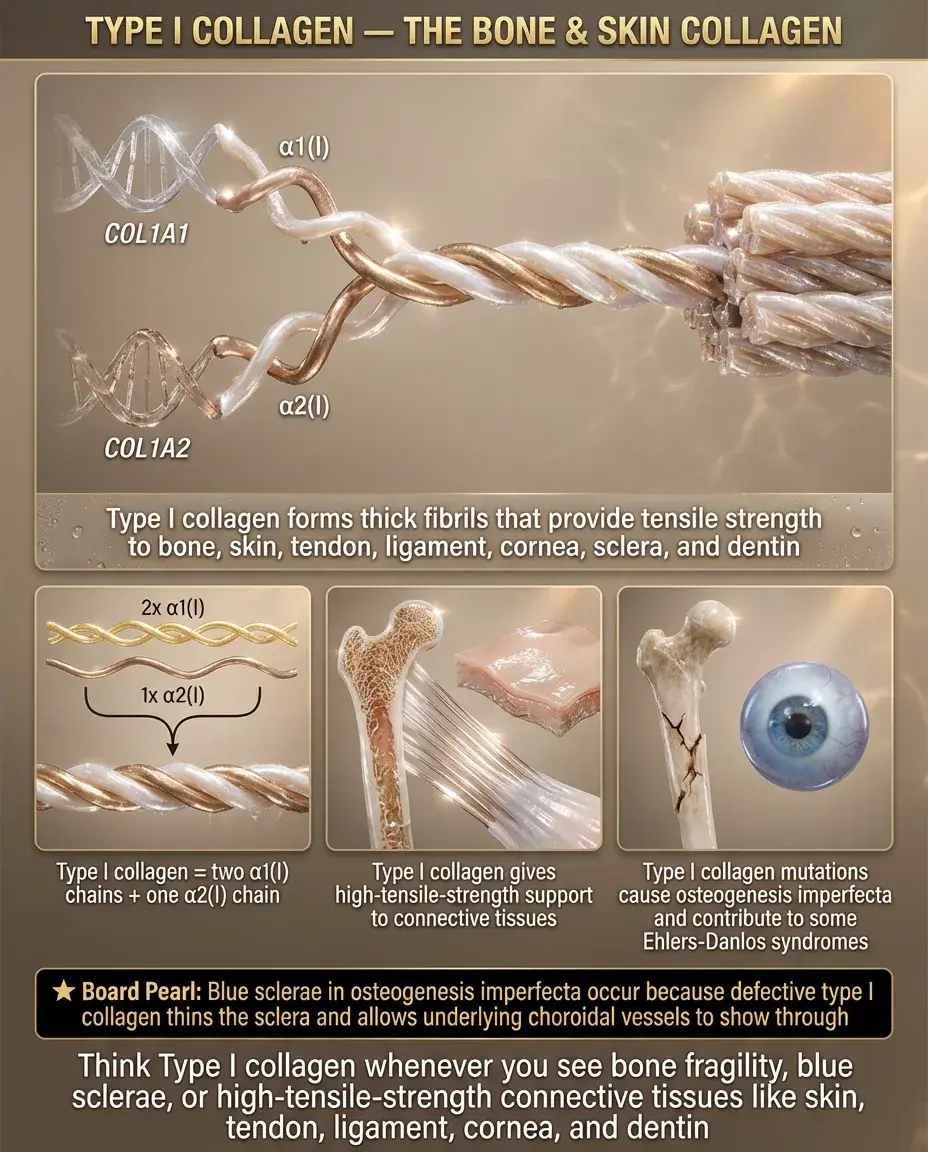
Type I Collagen: The Bone and Skin Collagen
Type I collagen (90% of body's collagen) forms thick fibrils providing tensile strength to bone, skin, tendons, ligaments, cornea, and dentin.
Composed of two α1(I) chains and one α2(I) chain encoded by COL1A1 and COL1A2 genes.
Mutations → osteogenesis imperfecta (brittle bones), with severity depending on whether the mutation causes reduced quantity (mild) or structurally abnormal collagen (severe).
Board pearl: Blue sclerae in OI result from thin sclera allowing underlying choroid vessels to show through — the sclera contains type I collagen.
Type I collagen is also defective in some forms of Ehlers-Danlos syndrome.
Type II Collagen: The Cartilage Collagen
Type II collagen is specific to cartilage (articular, costal, tracheal), vitreous humor, and nucleus pulposus.
Composed of three identical α1(II) chains forming thin fibrils that interact with proteoglycans to create the gel-like cartilage matrix.
Provides tensile strength while allowing reversible deformation — essential for joint function.
Mutations cause chondrodysplasias: skeletal malformations, short stature, early osteoarthritis.
Board distinction: Type I is in bone; Type II is in cartilage — remember "TWO" sounds like "TOO" as in carTOOlage.

Type III Collagen: The Reticular Collagen
Type III collagen forms thin, reticular fibers in skin, blood vessels, uterus, intestines, and granulation tissue — organs that need flexibility.
Composed of three α1(III) chains; often found alongside type I collagen but forms thinner fibrils.
Critical for blood vessel integrity and wound healing (appears early in granulation tissue, later replaced by type I).
Mutations → Ehlers-Danlos syndrome type IV (vascular type): arterial/bowel/uterine rupture, thin translucent skin, easy bruising.
Board pearl: Type III collagen defects → catastrophic vascular events in young adults (aortic dissection, organ rupture).

Type IV Collagen: The Basement Membrane Collagen
Type IV collagen forms sheet-like networks (not fibrils) that constitute the structural scaffold of all basement membranes.
Contains interruptions in the Gly-X-Y sequence allowing more flexibility; forms a chicken-wire meshwork via head-to-head and tail-to-tail associations.
Major component of glomerular basement membrane, alveolar basement membrane, and lens capsule.
Mutations → Alport syndrome: progressive nephritis, sensorineural deafness, ocular abnormalities (anterior lenticonus).
Anti-type IV collagen antibodies → Goodpasture syndrome (anti-GBM disease) targeting kidneys and lungs.
Board pearl: Basement membrane = Type IV (FOUR sounds like FLOOR).

Elastin and Elastic Fiber Assembly
Elastin provides tissues with elasticity — the ability to stretch and recoil (lungs, large arteries, skin, ligaments).
Composed of hydrophobic amino acids (glycine, valine, alanine, proline) with minimal hydroxyproline and no hydroxylysine or glycosylation.
Tropoelastin monomers are secreted and assembled on a scaffold of fibrillin microfibrils.
Cross-linking occurs via lysyl oxidase creating desmosine and isodesmosine — unique to elastin.
Defects in fibrillin-1 → Marfan syndrome; elastin gene deletion → Williams syndrome (supravalvular aortic stenosis, elfin facies).
α1-antitrypsin deficiency → unopposed elastase activity → emphysema.

Proteoglycans: The Shock Absorbers
Proteoglycans consist of a core protein covalently attached to glycosaminoglycan (GAG) chains — creating a bottle-brush structure.
GAGs are long, unbranched polysaccharides with repeating disaccharide units, highly negatively charged due to sulfate and carboxyl groups.
Negative charges attract cations and water → hydrated gel that resists compression (important in cartilage, vitreous humor).
Major proteoglycans: aggrecan (cartilage), versican (blood vessels), decorin (regulates collagen fibril assembly).
Board pearl: Proteoglycans provide the "bounce-back" in cartilage — compression squeezes out water, release allows rehydration.

Glycosaminoglycans and Their Clinical Correlates
Major GAGs: hyaluronic acid (not sulfated, not protein-linked), chondroitin sulfate, keratan sulfate, heparan sulfate, dermatan sulfate.
Lysosomal enzymes degrade GAGs; enzyme deficiencies → mucopolysaccharidoses with GAG accumulation.
Hurler syndrome (α-L-iduronidase deficiency): dermatan/heparan sulfate accumulation → coarse facies, corneal clouding, mental retardation.
Hunter syndrome (iduronate sulfatase deficiency): X-linked, similar to Hurler but no corneal clouding.
Board distinction: Hurler has corneal cLouding; Hunter (X-linked) patients are Hunters who need clear vision — no corneal clouding.

Fibronectin: The Cell-Matrix Connector
Fibronectin is a large glycoprotein that connects cells to the extracellular matrix via integrin receptors.
Contains binding sites for collagen, heparin, fibrin, and cells (via RGD sequence recognized by integrins).
Exists in two forms: plasma fibronectin (soluble, involved in wound healing) and cellular fibronectin (insoluble matrix component).
Critical for cell migration during embryogenesis, wound healing, and maintaining tissue architecture.
Forms fibrillar networks that guide cell movement and organize matrix assembly.
Board pearl: Fibronectin mediates cell adhesion and migration — important in wound healing and metastasis.

Laminin: The Basement Membrane Organizer
Laminin is a large cross-shaped glycoprotein found exclusively in basement membranes.
Forms networks by self-polymerization and binds to type IV collagen, nidogen/entactin, and cell surface receptors (integrins, dystroglycan).
Critical for basement membrane assembly and stability — without laminin, basement membranes cannot form.
Different laminin isoforms have tissue-specific distributions and functions.
Mutations → muscular dystrophies (especially affecting the neuromuscular junction) and epidermolysis bullosa.
Anti-laminin antibodies may contribute to some forms of pemphigoid.

Osteogenesis Imperfecta: When Bones Break
OI results from mutations in COL1A1 or COL1A2 genes encoding type I collagen α chains.
Type I (mild): reduced collagen quantity, normal structure → blue sclerae, hearing loss, mild bone fragility.
Type II (lethal): severe structural defects → death in utero or perinatally from respiratory failure.
Type III (severe deforming): progressively deforming, very short stature, triangular face.
Type IV (moderate): normal sclerae, moderate bone deformity.
Board pearl: Blue sclerae + multiple fractures + hearing loss = osteogenesis imperfecta until proven otherwise.

Ehlers-Danlos Syndromes: When Tissues Stretch
EDS comprises multiple disorders of collagen and collagen-modifying enzymes → joint hypermobility, skin hyperextensibility, tissue fragility.
Classical type (formerly I/II): type V collagen defect → hyperextensible skin, atrophic scars, joint hypermobility.
Vascular type (formerly IV): type III collagen defect → arterial/organ rupture, thin translucent skin, acrogeria.
Kyphoscoliotic type: lysyl hydroxylase deficiency → severe scoliosis, ocular fragility.
Board distinction: Vascular EDS is life-threatening; classical EDS causes problems but not catastrophic events.
Board clue: Young adult with spontaneous pneumothorax or arterial rupture → consider vascular EDS.

Marfan Syndrome: When Elastic Fibers Fail
Marfan syndrome results from mutations in FBN1 encoding fibrillin-1, which forms the scaffold for elastin deposition.
Without proper fibrillin microfibrils, elastic fibers are fragmented and tissues lose elasticity.
Clinical triad: skeletal (tall, arachnodactyly, pectus deformities), cardiovascular (aortic root dilation, mitral valve prolapse), ocular (lens dislocation).
Aortic dissection is the major cause of death — requires regular monitoring and prophylactic surgery.
Board pearl: Upward lens dislocation = Marfan; downward = homocystinuria.
Increased TGF-β signaling contributes to pathogenesis — basis for using losartan therapy.

Scurvy: When Collagen Can't Be Made
Vitamin C (ascorbic acid) is an essential cofactor for prolyl hydroxylase and lysyl hydroxylase → without it, collagen lacks hydroxyproline and hydroxylysine → unstable triple helix.
Clinical features reflect impaired collagen synthesis: bleeding gums, perifollicular hemorrhages, poor wound healing, bone pain.
Corkscrew hairs and perifollicular hemorrhages are pathognomonic.
In children: bone pain, refusal to walk, subperiosteal hemorrhages on X-ray.
Board pearl: Scurvy affects newly synthesized collagen — old collagen remains intact, explaining why teeth don't fall out in adults.
Treatment with vitamin C → rapid improvement within days.

Laboratory Diagnosis of Connective Tissue Disorders
Biochemical analysis: amino acid analysis of collagen shows decreased hydroxyproline in scurvy, abnormal cross-links in some EDS types.
Genetic testing: identifies specific mutations in collagen genes (OI, EDS), FBN1 (Marfan), enzyme genes (Hunter, Hurler).
Urine GAGs: elevated in mucopolysaccharidoses; specific patterns help differentiate types.
Skin biopsy: electron microscopy shows collagen fibril abnormalities in EDS, absent elastic fibers in cutis laxa.
Enzyme assays: for specific lysosomal storage diseases (Hunter, Hurler).
Board pearl: Genetic testing confirms diagnosis but clinical features often suffice for board questions.

Collagen Synthesis Steps and Potential Defects
Translation: procollagen α chains synthesized with signal peptide → ER.
Hydroxylation: prolyl and lysyl hydroxylase (require vitamin C, α-ketoglutarate, Fe²⁺) → deficiency causes scurvy.
Glycosylation: hydroxylysine residues glycosylated → defects in some EDS types.
Triple helix formation: three α chains assemble → glycine mutations cause OI.
Secretion and propeptide cleavage: defects → EDS type VII.
Cross-linking: lysyl oxidase (requires Cu²⁺) → deficiency causes Menkes disease.
Board pearl: Each step is a potential site for genetic or acquired defects.

Clinical Pattern Recognition
Blue sclerae + fractures + dentinogenesis imperfecta → osteogenesis imperfecta.
Hyperextensible skin + joint hypermobility + cigarette paper scars → classical Ehlers-Danlos.
Aortic dilation + lens dislocation + arachnodactyly → Marfan syndrome.
Arterial rupture + thin skin + acrogeria → vascular Ehlers-Danlos.
Bleeding gums + perifollicular hemorrhage + corkscrew hairs → scurvy.
Coarse facies + corneal clouding + developmental delay → Hurler syndrome.
Board strategy: Match the tissue affected to the protein likely defective — vascular problems suggest elastin or type III collagen.

Board Question Stem Patterns
Infant with multiple fractures and blue sclerae → osteogenesis imperfecta (type I collagen defect).
Young adult with aortic dissection and upward lens dislocation → Marfan syndrome (fibrillin-1 defect).
Patient with easy bruising and spontaneous bowel perforation → vascular Ehlers-Danlos (type III collagen defect).
Child refusing to walk with subperiosteal hemorrhages → scurvy (vitamin C deficiency).
Infant with corneal clouding and hepatosplenomegaly → mucopolysaccharidosis (Hurler syndrome).
Hyperextensible joints with normal skin → benign joint hypermobility vs EDS.
Positive thumb and wrist signs → screen for Marfan syndrome.

One-Line Recap
Connective tissue integrity depends on properly synthesized and assembled collagen types (I in bone/skin, II in cartilage, III in vessels, IV in basement membranes), elastin for stretch-recoil, proteoglycans for compression resistance, and adhesion molecules for cell-matrix connections — with defects causing predictable syndromes from brittle bones (OI) to vascular catastrophes (vascular EDS) to elastic fiber failure (Marfan).

bottom of page