top of page

eduo
visual

Hematology & Immunology
Mechanisms of Autoimmunity
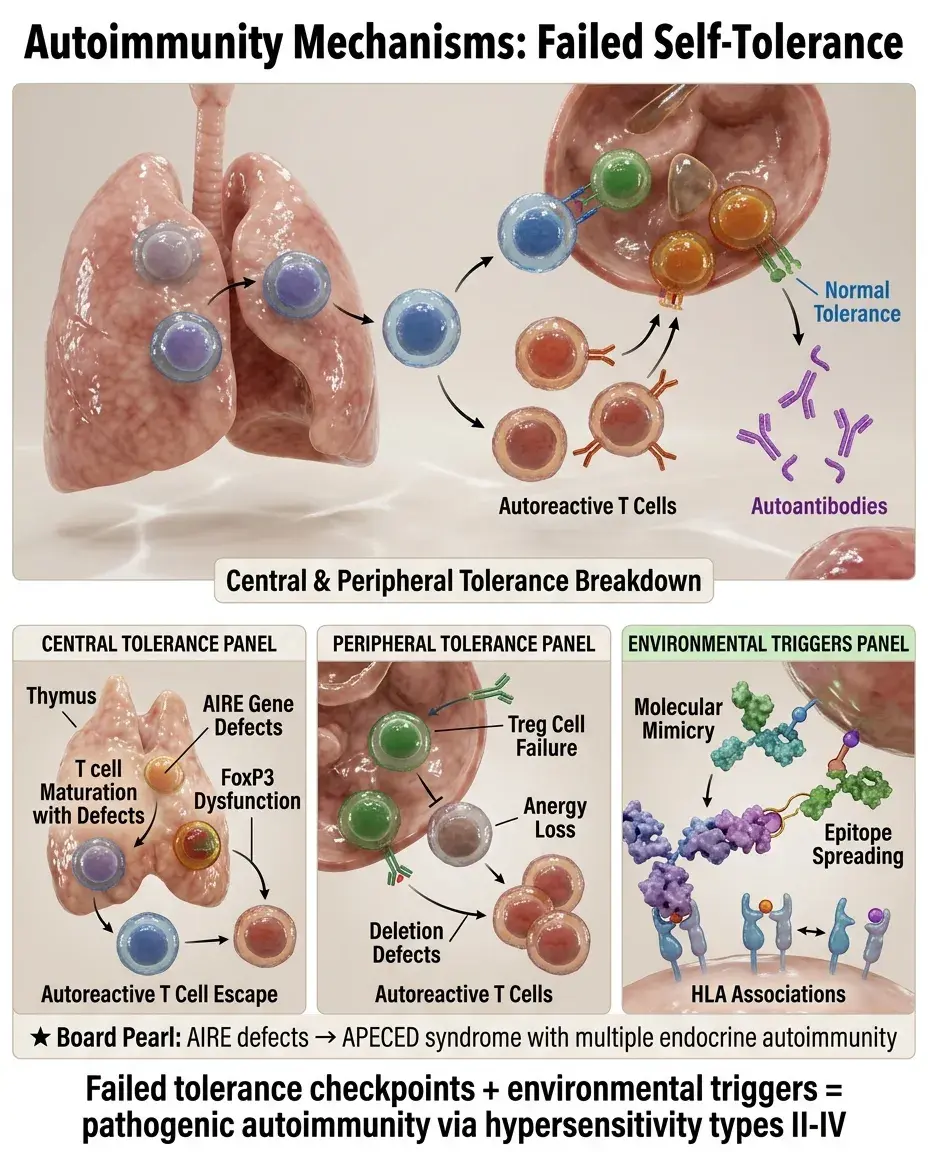
Core Principle of Autoimmunity
🧷
Autoimmunity occurs when the immune system fails to distinguish self from non-self, resulting in an attack on the body's own tissues.
🧷
This breakdown in self-tolerance involves both central tolerance (deletion of autoreactive T and B cells during development) and peripheral tolerance (regulatory mechanisms that suppress autoreactive cells that escape central deletion).
🧷
The development of autoimmune disease requires multiple hits: genetic susceptibility, environmental triggers, and failure of regulatory mechanisms.
🧷
Understanding these mechanisms explains why autoimmune diseases are chronic, why they often affect specific organs, and why they can be triggered by infections or other environmental factors.

Central Tolerance: The First Line of Defense
📍
Central tolerance occurs in primary lymphoid organs: thymus for T cells, bone marrow for B cells.
📍
In the thymus, T cells undergo positive selection (MHC recognition) followed by negative selection (deletion of cells recognizing self-antigens with high affinity).
📍
AIRE (autoimmune regulator) gene allows thymic epithelial cells to express tissue-restricted antigens, enabling deletion of T cells reactive to peripheral tissues.
📍
B cells undergo receptor editing in bone marrow — if BCR recognizes self-antigen, the cell attempts to rearrange a new light chain.
📍
Board pearl: Loss-of-function mutations in AIRE cause APECED syndrome (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy).

Peripheral Tolerance Mechanisms
🔹
Anergy: functional inactivation of lymphocytes that encounter antigen without proper costimulation (signal 1 without signal 2).
🔹
Deletion: activation-induced cell death via Fas-FasL pathway eliminates repeatedly stimulated self-reactive T cells.
🔹
Ignorance: physical separation of lymphocytes from self-antigens (blood-brain barrier, blood-testis barrier).
🔹
Suppression: regulatory T cells (Tregs) expressing CD4⁺CD25⁺FoxP3⁺ actively suppress autoreactive cells through IL-10, TGF-β, and cell contact-dependent mechanisms.
🔹
Board pearl: Loss of FoxP3 function causes IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked).

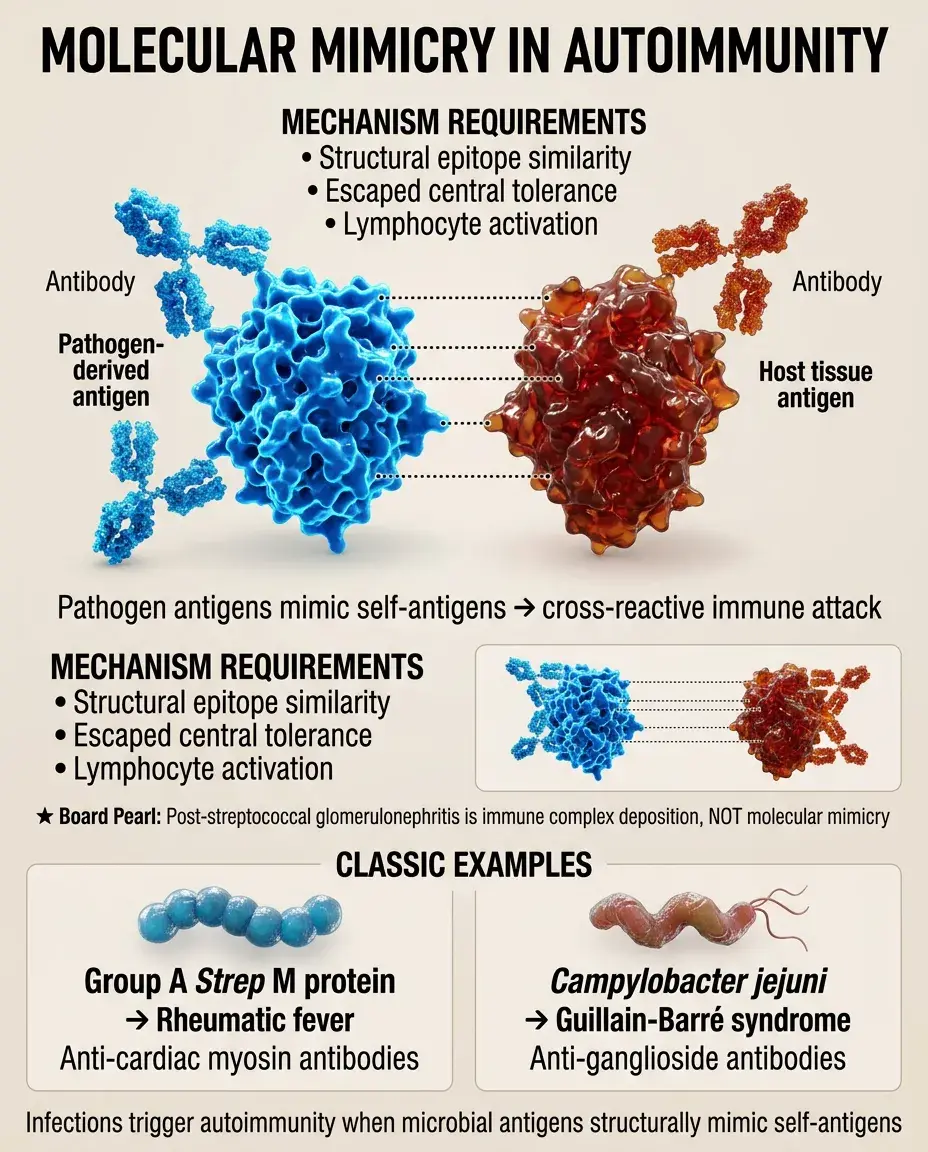
Molecular Mimicry: When Infections Trigger Autoimmunity
⭐
Molecular mimicry occurs when microbial antigens share structural similarity with self-antigens, leading to cross-reactive immune responses.
⭐
Classic examples: Group A Streptococcus M protein → rheumatic fever (anti-cardiac myosin antibodies); Campylobacter jejuni → Guillain-Barré syndrome (anti-ganglioside antibodies).
⭐
The immune response initially targets the pathogen but subsequently attacks host tissues expressing similar epitopes.
⭐
This mechanism requires both structural similarity and activation of autoreactive lymphocytes that escaped central tolerance.
⭐
Board pearl: Post-streptococcal glomerulonephritis is NOT molecular mimicry — it's immune complex deposition.
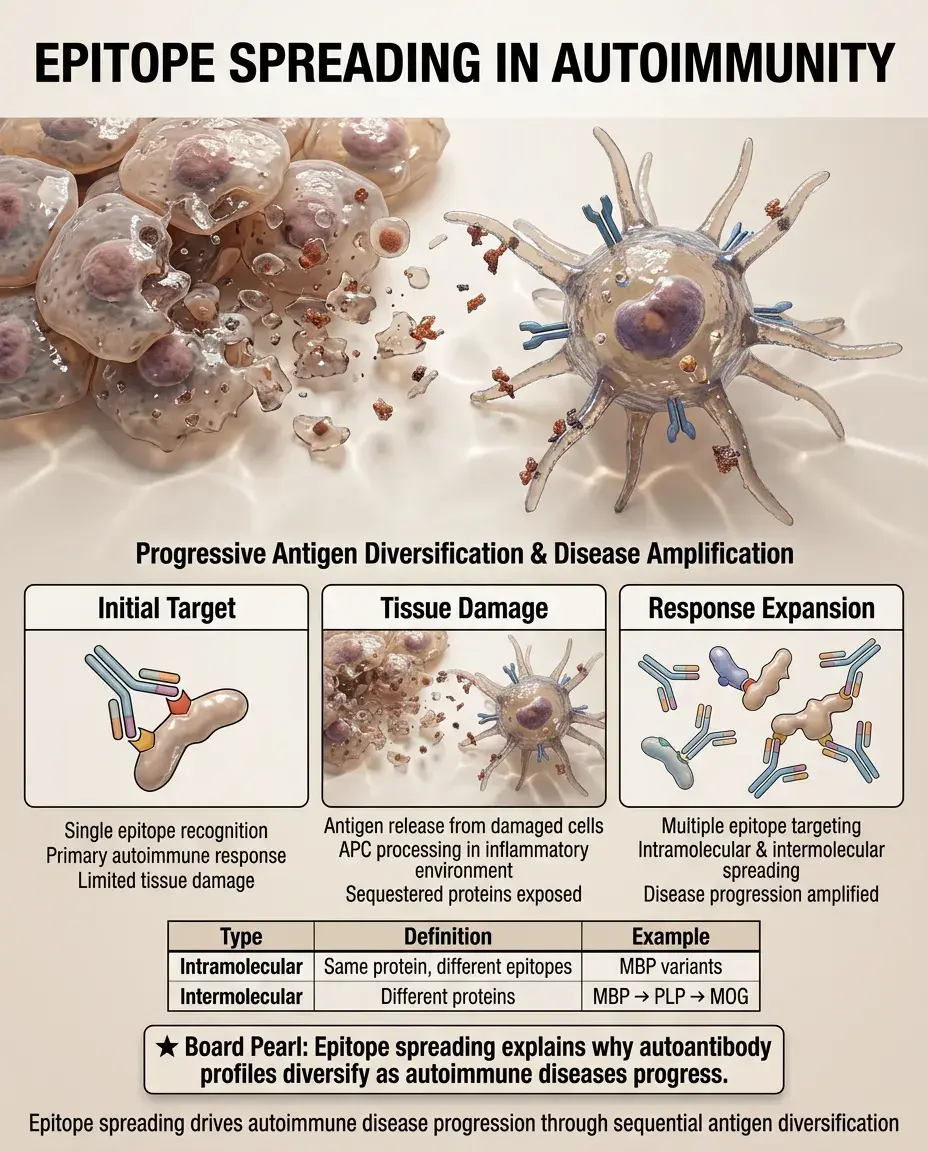
Epitope Spreading: Amplification of Autoimmunity
✅
Epitope spreading is the diversification of autoimmune response from initial target epitope to other epitopes on the same protein (intramolecular) or different proteins (intermolecular).
✅
Initial tissue damage releases previously sequestered antigens, which are processed and presented by APCs in an inflammatory environment.
✅
This process explains disease progression and why autoimmune responses broaden over time.
✅
Example: In multiple sclerosis, initial response to myelin basic protein can spread to proteolipid protein and myelin oligodendrocyte glycoprotein.
✅
Board clue: Epitope spreading explains why autoantibody profiles become more diverse as disease progresses.
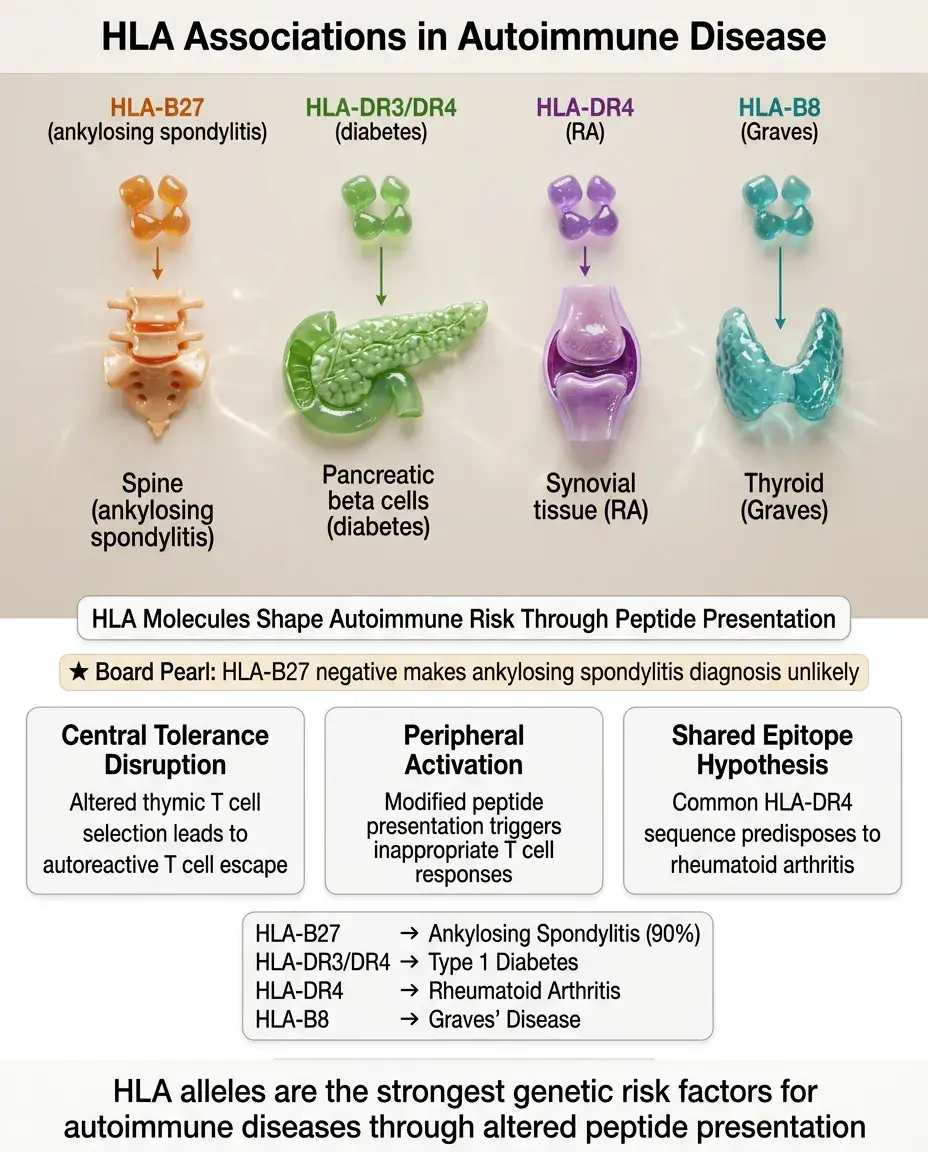
HLA Associations and Genetic Susceptibility
🧠
Specific HLA alleles are the strongest genetic risk factors for most autoimmune diseases.
🧠
HLA-B27: ankylosing spondylitis (90% association), reactive arthritis, psoriatic arthritis.
🧠
HLA-DR3/DR4: type 1 diabetes (especially DQ2/DQ8 haplotypes).
🧠
HLA-DR4: rheumatoid arthritis (shared epitope hypothesis).
🧠
HLA-B8: Graves' disease, myasthenia gravis.
🧠
The mechanism involves altered peptide presentation, affecting both central tolerance (thymic selection) and peripheral T cell activation.
🧠
Board pearl: HLA-B27 is so strongly associated with ankylosing spondylitis that its absence makes the diagnosis unlikely.
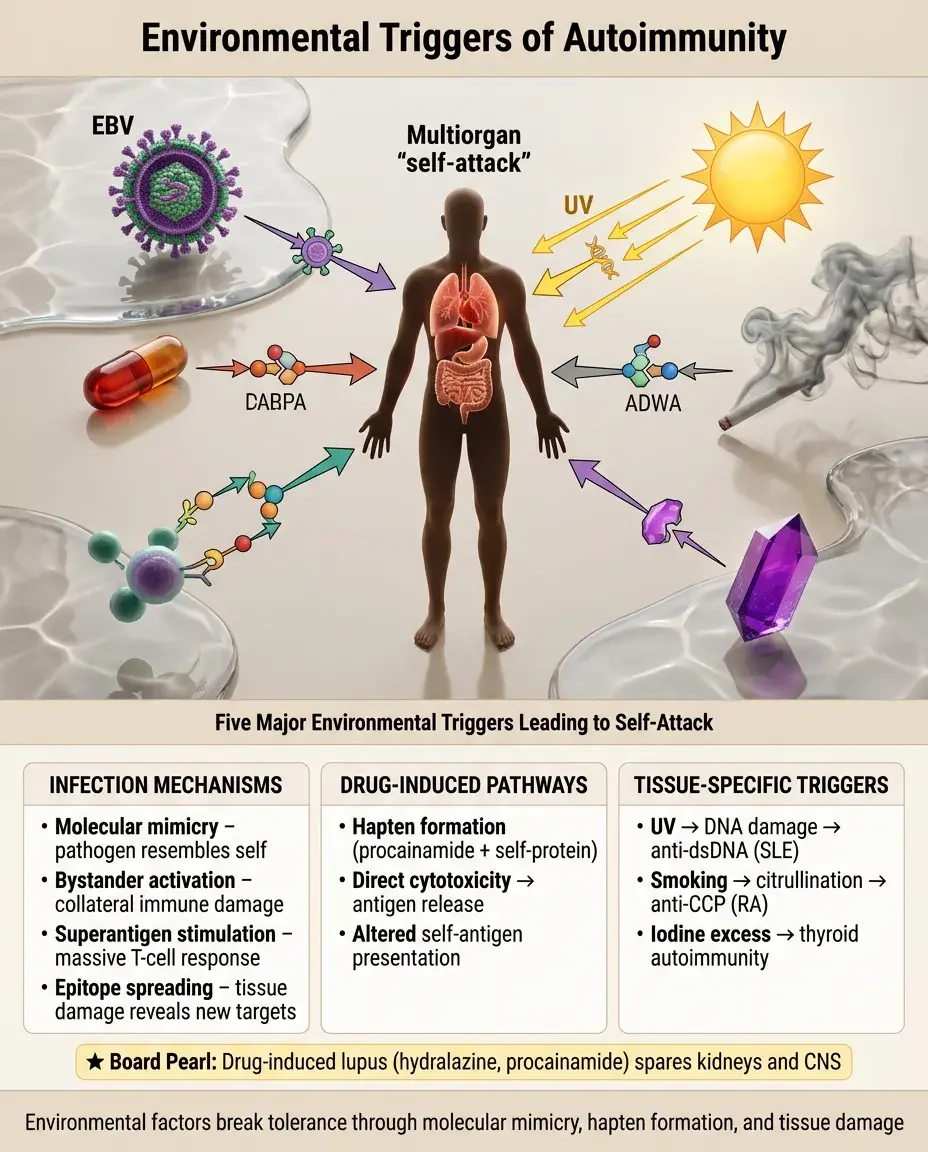
Environmental Triggers of Autoimmunity
⚡
Infections: molecular mimicry, bystander activation, superantigen stimulation, epitope spreading from tissue damage.
⚡
Drugs: can act as haptens (procainamide → lupus-like syndrome), alter self-antigens, or cause direct cytotoxicity with antigen release.
⚡
UV radiation: causes DNA damage and apoptosis → release of nuclear antigens → anti-dsDNA antibodies in SLE.
⚡
Smoking: citrullination of proteins in lungs → anti-CCP antibodies in rheumatoid arthritis.
⚡
Iodine excess: can precipitate thyroid autoimmunity in genetically susceptible individuals.
⚡
Board pearl: Drug-induced lupus (hydralazine, procainamide) typically spares kidneys and CNS.
Defective Apoptosis and Autoantigen Persistence
📌
Normal apoptosis includes phosphatidylserine externalization, promoting anti-inflammatory clearance by phagocytes.
📌
Defective clearance of apoptotic cells leads to secondary necrosis → release of intracellular antigens in pro-inflammatory context.
📌
C1q deficiency: impaired clearance of immune complexes and apoptotic cells → severe early-onset SLE.
📌
Fas/FasL mutations: defective activation-induced cell death → autoimmune lymphoproliferative syndrome (ALPS).
📌
DNase mutations: impaired degradation of chromatin from dead cells → anti-nuclear antibodies.
📌
Board pearl: Complete C1q deficiency has >90% penetrance for SLE-like disease.

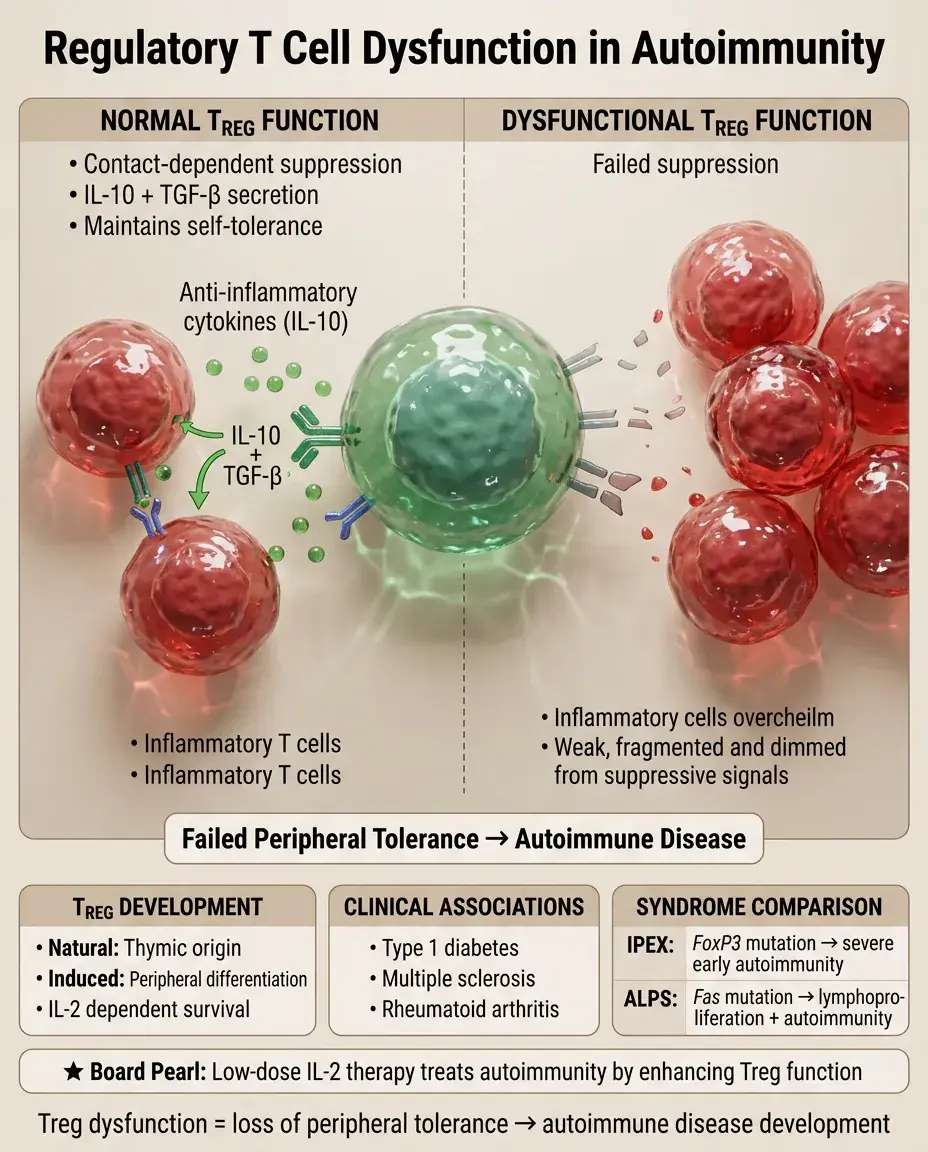
Regulatory T Cell Dysfunction
📣
Tregs (CD4⁺CD25⁺FoxP3⁺) maintain peripheral tolerance through contact-dependent suppression and anti-inflammatory cytokines (IL-10, TGF-β).
📣
Natural Tregs develop in thymus; induced Tregs differentiate in periphery from conventional CD4⁺ T cells.
📣
Treg deficiency or dysfunction is implicated in multiple autoimmune diseases: type 1 diabetes, multiple sclerosis, rheumatoid arthritis.
📣
IL-2 is essential for Treg survival and function — explains why low-dose IL-2 therapy can paradoxically treat autoimmunity.
📣
Board distinction: IPEX syndrome (FoxP3 mutation) → early severe autoimmunity; ALPS (Fas mutation) → lymphoproliferation with autoimmunity.
B Cell Tolerance Breakdown
🔸
Central B cell tolerance: receptor editing, clonal deletion, anergy induction in bone marrow.
🔸
Peripheral checkpoints: anergy maintenance, exclusion from follicles, lack of T cell help, FcγRIIB-mediated suppression.
🔸
Autoreactive B cells can be rescued by: strong BCR signaling, TLR co-stimulation, excessive BAFF (B cell activating factor).
🔸
Somatic hypermutation in germinal centers can generate de novo autoreactive B cells from non-autoreactive precursors.
🔸
Board pearl: Belimumab (anti-BAFF) treats SLE by reducing survival signals for autoreactive B cells.

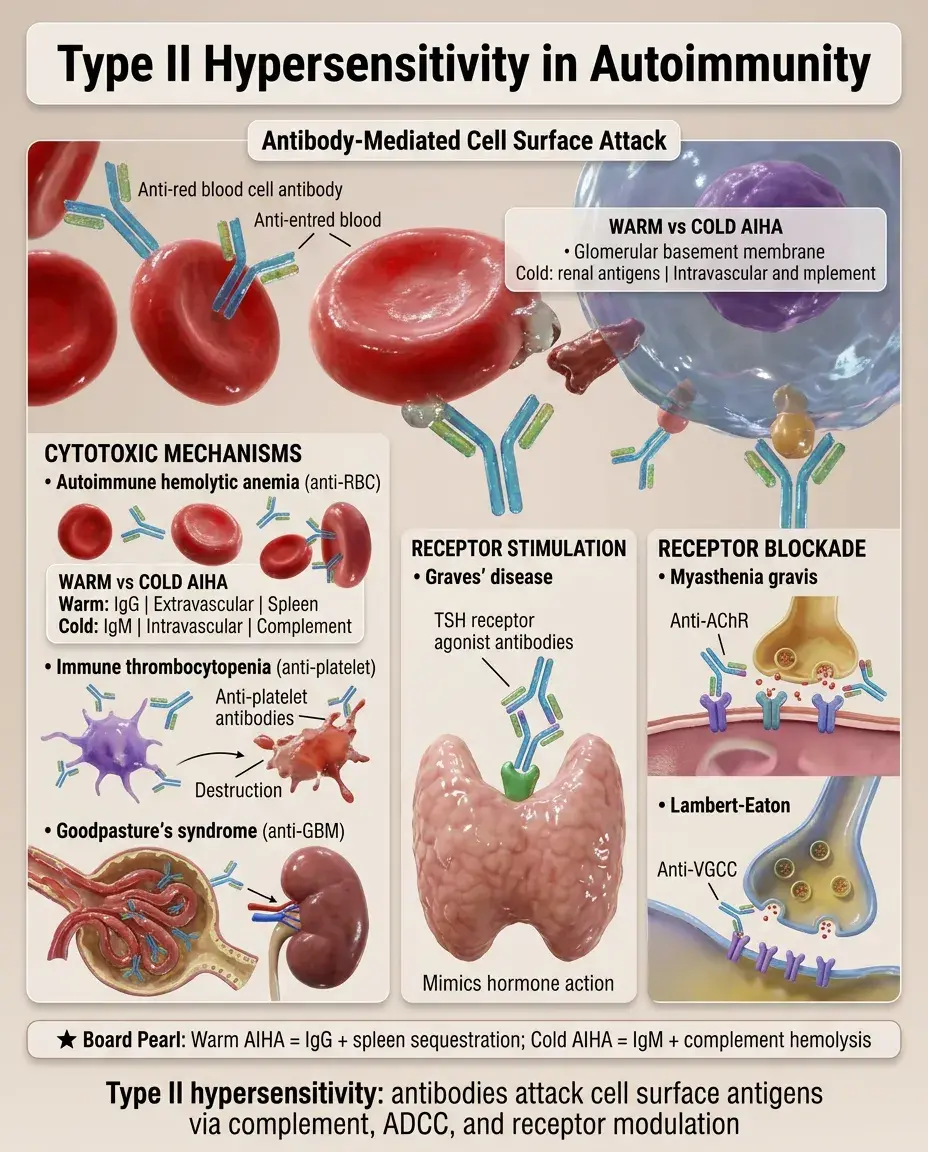
Type II Hypersensitivity in Autoimmunity
🧷
Antibodies bind cell surface antigens → complement activation, ADCC, opsonization, receptor blockade/stimulation.
🧷
Cytotoxic examples: autoimmune hemolytic anemia (anti-RBC), immune thrombocytopenia (anti-platelet), Goodpasture's (anti-GBM).
🧷
Receptor stimulation: Graves' disease (TSH receptor agonist antibodies).
🧷
Receptor blockade: myasthenia gravis (anti-AChR), Lambert-Eaton (anti-VGCC).
🧷
Board distinction: Warm AIHA (IgG, extravascular hemolysis in spleen) vs Cold AIHA (IgM, complement-mediated intravascular hemolysis).
Type III Hypersensitivity in Autoimmunity
📍
Immune complex formation → complement activation → neutrophil recruitment → tissue damage.
📍
Circulating complexes: SLE (anti-dsDNA/DNA complexes), cryoglobulinemia, rheumatoid arthritis (rheumatoid factor/IgG complexes).
📍
In situ formation: poststreptococcal glomerulonephritis (planted antigens).
📍
Factors favoring pathogenic complex formation: antigen excess, intermediate-sized complexes, impaired clearance mechanisms.
📍
Board pearl: Serum sickness-like reactions occur 7-10 days after antigen exposure, when antibody production reaches levels forming pathogenic complexes.

Type IV Hypersensitivity in Autoimmunity
🔹
T cell-mediated tissue damage through cytotoxic T cells or inflammatory cytokines from Th1/Th17 cells.
🔹
Direct cytotoxicity: type 1 diabetes (CD8⁺ T cells destroy β cells), autoimmune hepatitis (hepatocyte destruction).
🔹
Inflammatory damage: multiple sclerosis (Th1/Th17 attack myelin), rheumatoid arthritis (synovial inflammation).
🔹
Granuloma formation: sarcoidosis, Crohn's disease (though infectious causes must be excluded).
🔹
Board pearl: The tuberculin skin test is the classic example of type IV hypersensitivity — peaks at 48-72 hours.

Cytokine Networks in Autoimmunity
⭐
Th1 diseases (IL-12/IFN-γ axis): type 1 diabetes, multiple sclerosis, Hashimoto's thyroiditis.
⭐
Th2 diseases (IL-4/IL-13 axis): less common in autoimmunity, some allergic diseases with autoimmune features.
⭐
Th17 diseases (IL-23/IL-17 axis): psoriasis, ankylosing spondylitis, inflammatory bowel disease.
⭐
Type I interferons: central to SLE pathogenesis (plasmacytoid dendritic cells produce IFN-α).
⭐
TNF-α: key inflammatory mediator in rheumatoid arthritis, inflammatory bowel disease, psoriasis.
⭐
Board pearl: Anti-TNF therapy can paradoxically trigger demyelination or lupus-like syndrome.

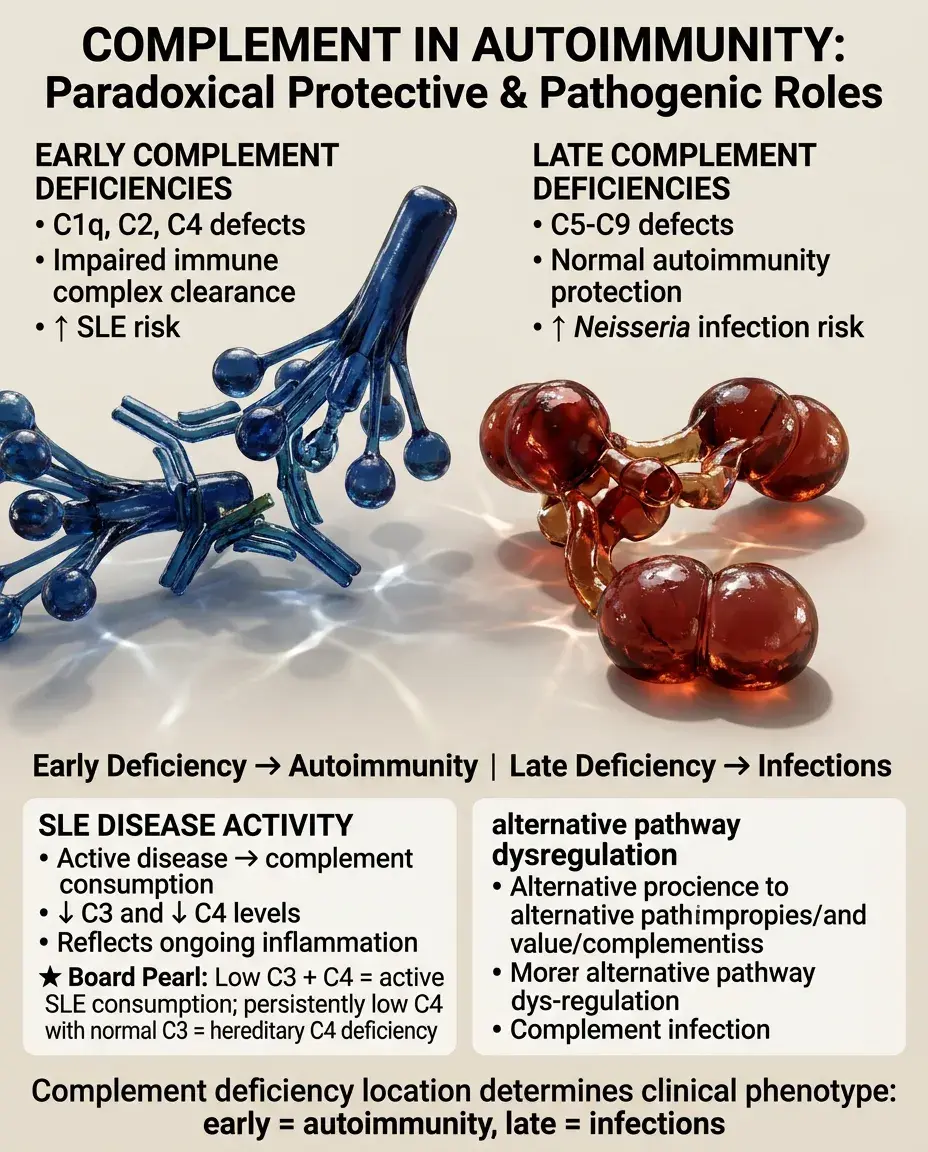
Complement in Autoimmunity: Paradoxical Roles
✅
Early complement deficiencies (C1q, C2, C4) → increased SLE risk due to impaired immune complex clearance.
✅
Late complement deficiencies (C5-C9) → increased risk of Neisseria infections but not autoimmunity.
✅
Complement activation in active disease: consumption leads to low C3, C4 levels (SLE disease activity marker).
✅
Alternative pathway dysregulation: C3 nephritic factor in membranoproliferative glomerulonephritis.
✅
Board pearl: Low C3 and C4 in active SLE reflects consumption; persistently low C4 with normal C3 may indicate hereditary deficiency.
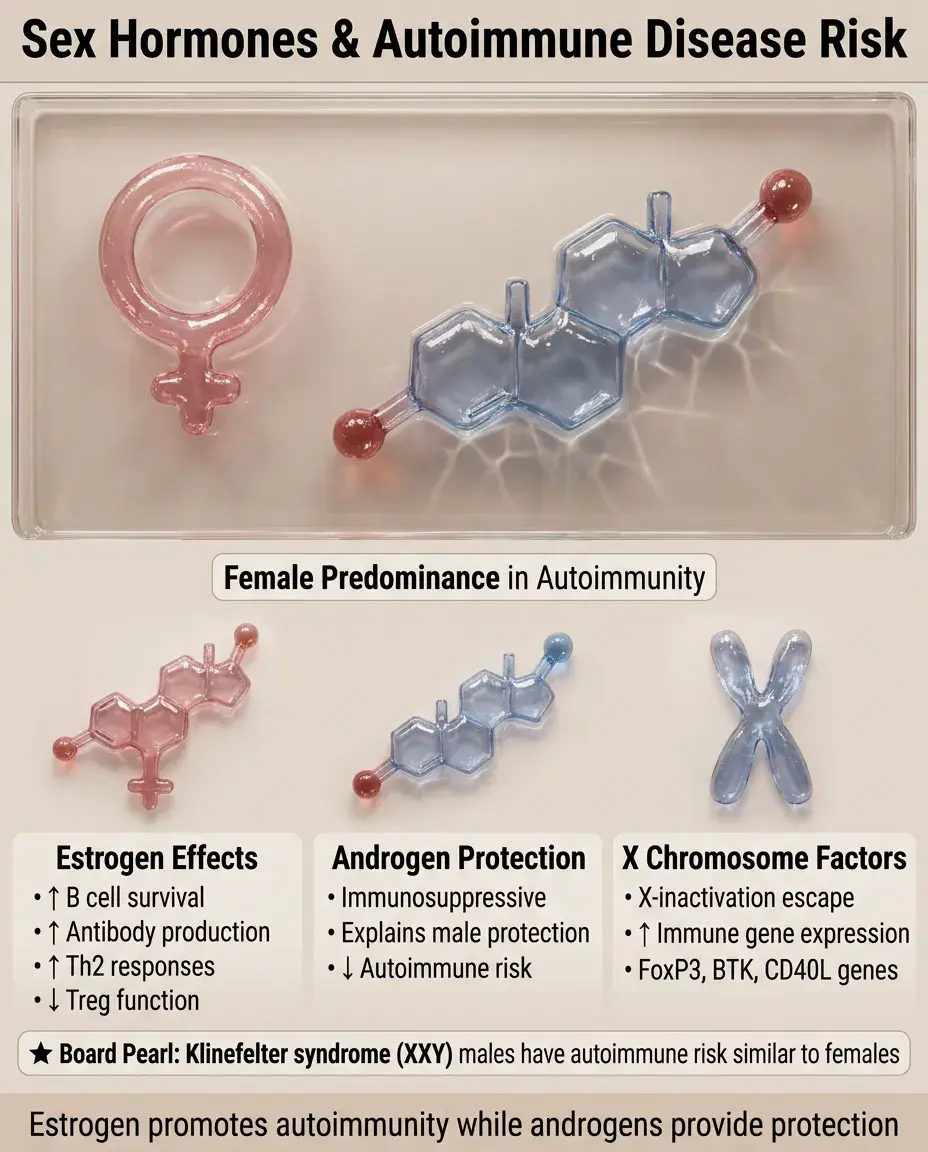
Sex Hormones and Autoimmunity
🧠
Female predominance in most autoimmune diseases: SLE (9:1), Sjögren's (9:1), autoimmune thyroid disease (5-8:1).
🧠
Estrogen effects: enhances B cell survival, increases antibody production, promotes Th2 responses, inhibits Treg function.
🧠
Androgen effects: generally immunosuppressive, may explain male protection.
🧠
X chromosome factors: X-inactivation escape leads to higher expression of immune genes; X-linked genes include FoxP3, BTK, CD40L.
🧠
Pregnancy effects: Th2 shift improves Th1 diseases (MS, RA) but may worsen SLE.
🧠
Board pearl: Klinefelter syndrome (XXY) males have increased autoimmune disease risk similar to females.
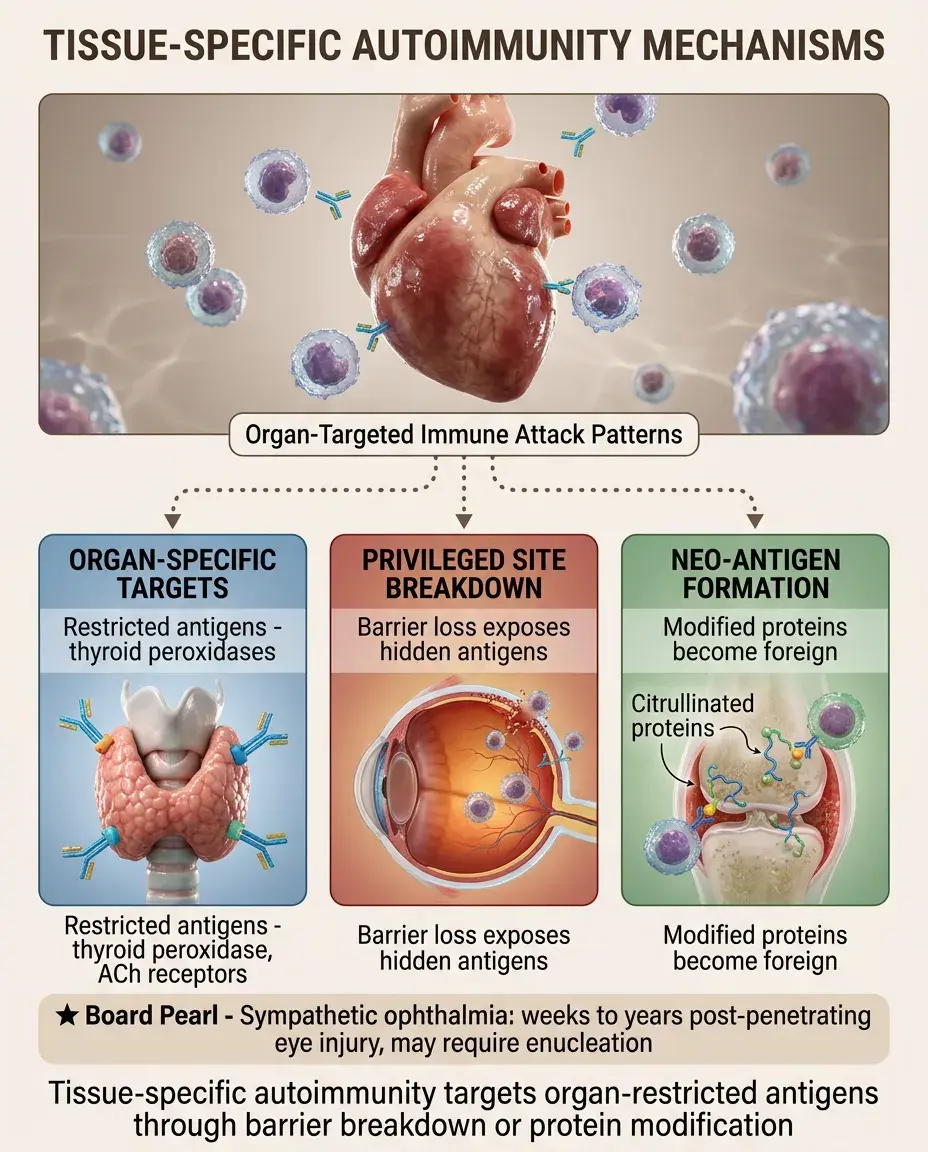
Tissue-Specific Autoimmunity Mechanisms
⚡
Organ-specific: antigenic targets restricted to particular tissues (thyroid peroxidase in Hashimoto's, acetylcholine receptor in myasthenia).
⚡
Immunologically privileged sites: when barriers break down (sympathetic ophthalmia after eye trauma, orchitis after vasectomy).
⚡
Neo-antigen formation: tissue-specific post-translational modifications (citrullination in RA, β cell proteins in diabetes).
⚡
Local factors: tissue-specific expression of costimulatory molecules, cytokines, or antigen-presenting cells.
⚡
Board pearl: Sympathetic ophthalmia can occur weeks to years after penetrating eye injury — requires enucleation consideration.
Therapeutic Principles Based on Mechanisms
📌
Immunosuppression: corticosteroids (broad), methotrexate (antifolate), azathioprine (purine synthesis inhibition).
📌
B cell depletion: rituximab (anti-CD20) removes antibody-producing cell precursors.
📌
T cell modulation: abatacept (CTLA-4-Ig) blocks costimulation, cyclosporine/tacrolimus inhibit calcineurin.
📌
Cytokine targeting: TNF inhibitors, IL-6 blockade (tocilizumab), IL-17 inhibitors, JAK inhibitors.
📌
Complement inhibition: eculizumab (anti-C5) for diseases with MAC-mediated damage.
📌
Board pearl: Rituximab spares plasma cells (CD20-negative), so autoantibody levels may remain elevated initially.

Board Question Stem Patterns
📣
Young woman with malar rash and proteinuria → SLE with immune complex glomerulonephritis.
📣
Positive ANA with negative anti-dsDNA and anti-Sm → consider drug-induced lupus or other CTDs.
📣
Type 1 diabetic developing another autoimmune disease → autoimmune polyglandular syndrome.
📣
Recurrent Neisseria infections → terminal complement deficiency.
📣
Chronic diarrhea in infant boy with eczema → IPEX syndrome (FoxP3 mutation).
📣
Post-infectious ascending paralysis → molecular mimicry (Guillain-Barré).
📣
Low C3, normal C4 → alternative pathway activation (C3 nephritic factor, factor H deficiency).

One-Line Recap
🔸
Autoimmunity results from failed self-tolerance through defects in central tolerance (AIRE, FoxP3), peripheral tolerance (Tregs, anergy, deletion), environmental triggers (molecular mimicry, epitope spreading), genetic susceptibility (HLA associations), creating pathogenic autoreactive T cells, autoantibodies, and immune complexes that drive organ-specific or systemic disease through type II, III, and IV hypersensitivity mechanisms.
bottom of page