top of page

eduo
visual

Reproductive & Endocrine Systems
Insulin synthesis, secretion, and signaling
Core Principle of Insulin Biology
🧷
Insulin is the primary anabolic hormone, synthesized by pancreatic β cells in response to nutrients (especially glucose) and secreted to promote energy storage.
🧷
The insulin signaling cascade begins with receptor autophosphorylation and culminates in glucose uptake, glycogen synthesis, lipogenesis, and protein synthesis while suppressing gluconeogenesis, glycogenolysis, and lipolysis.
🧷
Understanding the molecular details of insulin synthesis → secretion → receptor signaling → metabolic effects is essential for comprehending diabetes pathophysiology and drug mechanisms.
🧷
Board pearl: Insulin is the only hormone that lowers blood glucose; all counter-regulatory hormones (glucagon, cortisol, growth hormone, epinephrine) raise it.

Insulin Gene Structure and Transcription
📍
The INS gene on chromosome 11p15.5 encodes preproinsulin, regulated by glucose-responsive transcription factors including PDX-1, MafA, and NeuroD1.
📍
Glucose stimulates insulin gene transcription through metabolic signals that activate these β-cell-specific transcription factors, creating a feed-forward amplification.
📍
Mutations in transcription factors (especially PDX-1) cause maturity-onset diabetes of the young (MODY), characterized by β-cell dysfunction without insulin resistance.
📍
The insulin gene contains a variable number tandem repeat (VNTR) region upstream; class I alleles are associated with type 1 diabetes susceptibility.
📍
Board pearl: MODY presents as non-insulin-dependent diabetes in patients <25 years old with strong family history — think transcription factor mutations.

Preproinsulin Processing in the Endoplasmic Reticulum
🔹
Preproinsulin (110 amino acids) contains an N-terminal signal peptide that directs it to the rough ER for co-translational translocation.
🔹
Signal peptidase cleaves the 24-amino acid signal sequence during translation, yielding proinsulin (86 amino acids).
🔹
In the ER lumen, proinsulin folds with formation of three disulfide bonds: A7-B7, A20-B19, and A6-A11 (two interchain, one intrachain).
🔹
Proper disulfide bond formation is critical — misfolded proinsulin triggers ER stress and β-cell apoptosis, contributing to diabetes pathogenesis.
🔹
Board pearl: ER stress from chronic insulin demand (insulin resistance) leads to β-cell failure — the transition from prediabetes to type 2 diabetes.

Proinsulin Cleavage in Secretory Granules
⭐
Proinsulin is packaged into immature secretory granules with prohormone convertases PC1/3 and PC2, carboxypeptidase E, and acidic pH.
⭐
PC1/3 cleaves at the B-chain/C-peptide junction (Arg31-Arg32), while PC2 cleaves at the C-peptide/A-chain junction (Lys64-Arg65).
⭐
Carboxypeptidase E removes the exposed basic residues, yielding mature insulin (51 amino acids: 21-residue A chain + 30-residue B chain) and C-peptide (31 amino acids).
⭐
Insulin and C-peptide are stored in equimolar amounts in mature granules as crystalline hexamers coordinated by Zn²⁺.
⭐
Board pearl: C-peptide levels distinguish endogenous insulin production (present) from exogenous insulin administration (absent) — key for diagnosing factitious hypoglycemia.

Glucose Sensing and Metabolism in β Cells
✅
GLUT2 (Km ~15-20 mM) allows glucose entry proportional to blood glucose, functioning as the glucose sensor.
✅
Glucokinase (hexokinase IV, Km ~10 mM) phosphorylates glucose to G6P, serving as the rate-limiting step and metabolic gatekeeper.
✅
Glucose metabolism through glycolysis and the TCA cycle generates ATP, increasing the ATP/ADP ratio — this metabolic signal couples glucose sensing to insulin secretion.
✅
Glucokinase mutations cause MODY2 (mild hyperglycemia) when heterozygous or permanent neonatal diabetes when homozygous.
✅
Board pearl: Sulfonylureas bypass glucose sensing by directly closing KATP channels, explaining why they cause hypoglycemia even when glucose is low.

The KATP Channel and Membrane Depolarization
🧠
The ATP-sensitive potassium channel consists of four Kir6.2 pore subunits and four SUR1 regulatory subunits.
🧠
Rising ATP/ADP ratio closes KATP channels by ATP binding to Kir6.2, while ADP binding to SUR1 opposes closure.
🧠
KATP channel closure prevents K⁺ efflux → membrane depolarization from −70 mV to above −40 mV → voltage-gated Ca²⁺ channel opening.
🧠
Sulfonylureas bind SUR1 to close KATP channels independent of glucose; diazoxide opens channels to inhibit insulin secretion.
🧠
Board pearl: Congenital hyperinsulinism results from KATP channel mutations causing unregulated insulin secretion → severe neonatal hypoglycemia requiring diazoxide or pancreatectomy.

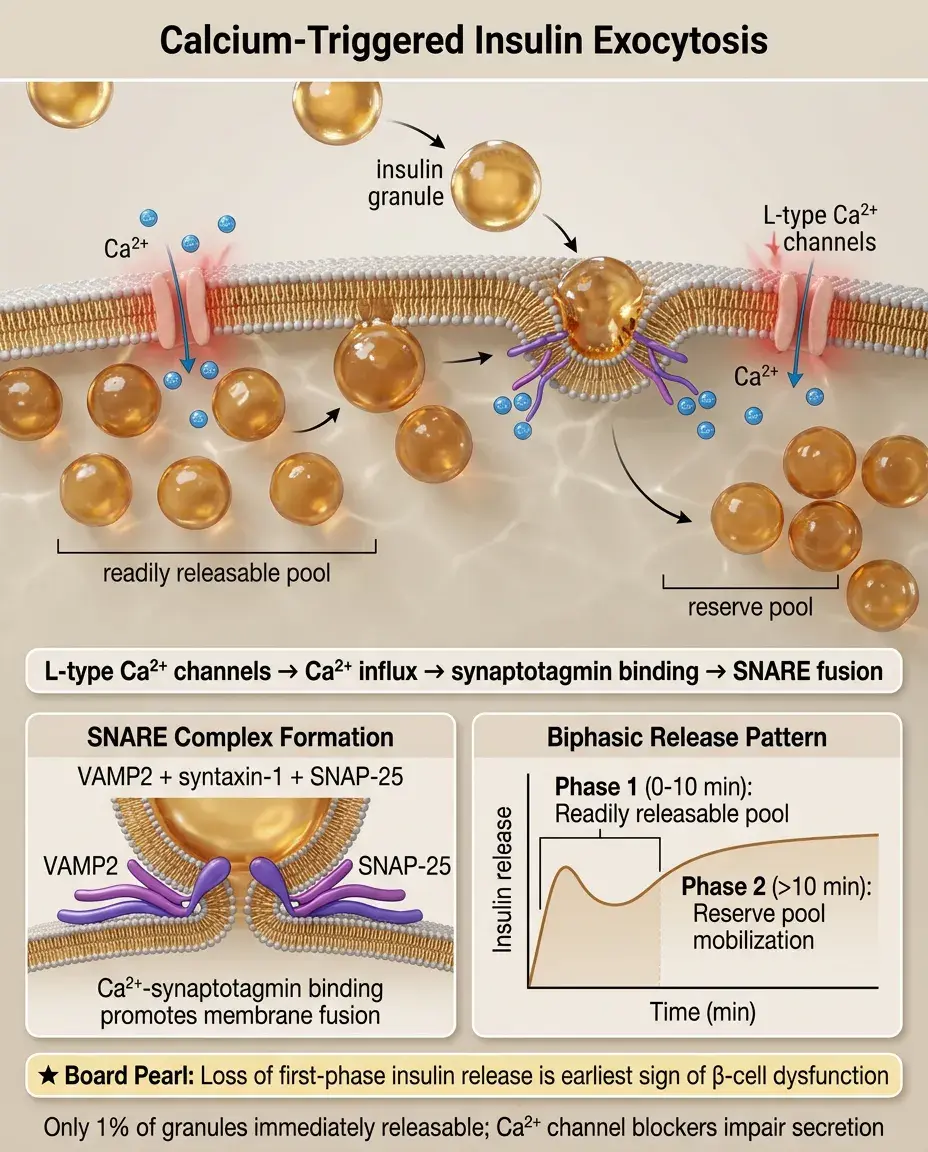
Calcium Influx and Exocytosis
⚡
Depolarization opens L-type voltage-gated Ca²⁺ channels (Cav1.2/1.3), causing rapid Ca²⁺ influx that triggers exocytosis.
⚡
Ca²⁺ binds synaptotagmin on insulin granules, promoting SNARE complex formation (VAMP2, syntaxin-1, SNAP-25) and membrane fusion.
⚡
Only ~1% of insulin granules are immediately releasable; the rest require mobilization from reserve pools through Ca²⁺-dependent cytoskeletal remodeling.
⚡
Calcium channel blockers can impair insulin secretion, while calcium channel activators (BAY K 8644) enhance it.
⚡
Board pearl: First-phase insulin release (0-10 minutes) empties the readily releasable pool; second-phase (>10 minutes) requires granule mobilization — loss of first-phase is the earliest sign of β-cell dysfunction.
Insulin Receptor Structure and Activation
📌
The insulin receptor is a heterotetrameric receptor tyrosine kinase: two extracellular α subunits (insulin binding) linked by disulfide bonds to two transmembrane β subunits (kinase domains).
📌
Insulin binding to the α subunits causes conformational change → trans-autophosphorylation of β subunit tyrosines (Y1158, Y1162, Y1163 in the activation loop).
📌
Activated receptor phosphorylates insulin receptor substrates (IRS-1 through IRS-4) on multiple tyrosines, creating docking sites for SH2-domain proteins.
📌
Receptor internalization and degradation regulate signaling duration; hyperinsulinemia accelerates receptor downregulation.
📌
Board pearl: Insulin receptor antibodies can cause severe insulin resistance (type B insulin resistance) or paradoxical hypoglycemia by acting as agonists.

PI3K-AKT Pathway: Metabolic Signaling
📣
IRS tyrosine phosphorylation recruits PI3K via SH2 domains → PI3K phosphorylates PIP2 to PIP3 → PDK1 and mTORC2 activate AKT.
📣
AKT phosphorylates AS160 (TBC1D4) → Rab-GTP accumulation → GLUT4 vesicle translocation to plasma membrane → glucose uptake in muscle/adipose.
📣
AKT phosphorylates and inactivates GSK3β → glycogen synthase activation → glycogen synthesis.
📣
AKT phosphorylates and inactivates FOXO1 → suppression of gluconeogenic genes (PEPCK, G6Pase) in liver.
📣
Board pearl: Metformin primarily works through AMPK activation, not insulin signaling, explaining why it doesn't cause hypoglycemia as monotherapy.

MAPK Pathway: Mitogenic Signaling
🔸
IRS phosphorylation also recruits Grb2-SOS → Ras activation → RAF → MEK → ERK cascade.
🔸
ERK translocates to nucleus → phosphorylates transcription factors (Elk-1, c-Myc) → cell proliferation and differentiation.
🔸
The MAPK pathway mediates insulin's growth-promoting effects, explaining the association between hyperinsulinemia and cancer risk.
🔸
Selective insulin resistance preserves MAPK signaling while metabolic (PI3K-AKT) signaling is impaired, contributing to diabetic complications.
🔸
Board pearl: IGF-1 shares downstream signaling with insulin but has greater mitogenic potential — this explains why insulin analogues with increased IGF-1 receptor affinity raised cancer concerns.

Tissue-Specific Insulin Actions
🧷
Muscle (40% of body mass): GLUT4 translocation → glucose uptake for glycogen synthesis and oxidation. Primary site of postprandial glucose disposal.
🧷
Adipose: GLUT4 translocation → glucose uptake for triglyceride synthesis. LPL activation → fatty acid uptake. HSL inhibition → antilipolysis.
🧷
Liver: No GLUT4; insulin suppresses glucose output by inhibiting gluconeogenesis/glycogenolysis and promotes glycogen synthesis and de novo lipogenesis.
🧷
Brain: Largely insulin-independent glucose uptake via GLUT1/3, but insulin affects satiety, reward pathways, and cognitive function.
🧷
Board pearl: Muscle insulin resistance appears first in type 2 diabetes progression, followed by adipose, then hepatic resistance.

Molecular Mechanisms of Insulin Resistance
📍
Serine phosphorylation of IRS proteins by JNK, IKKβ, PKCθ, and S6K impairs tyrosine phosphorylation → reduced PI3K activation.
📍
Lipid metabolites (ceramides, diacylglycerol) activate PKC isoforms → serine phosphorylation of insulin signaling proteins.
📍
Inflammatory cytokines (TNF-α, IL-6) activate serine kinases and induce SOCS proteins that promote IRS degradation.
📍
ER stress → unfolded protein response → JNK activation → IRS serine phosphorylation.
📍
Board pearl: The common pathway in obesity-induced insulin resistance is ectopic lipid accumulation → lipotoxicity → inflammatory signaling → IRS dysfunction.

Regulation of Insulin Secretion
🔹
Glucose is the primary stimulus, but amino acids (especially leucine, arginine), fatty acids, and hormones modulate the response.
🔹
Incretins (GLP-1, GIP) amplify glucose-stimulated insulin secretion via cAMP-PKA and EPAC2 pathways — explaining why oral glucose causes greater insulin release than IV glucose.
🔹
Autonomic innervation: vagal (cholinergic) stimulation enhances secretion via M3 receptors; sympathetic (adrenergic) inhibits via α2 receptors.
🔹
Paracrine regulation: α-cell glucagon stimulates, δ-cell somatostatin inhibits insulin secretion.
🔹
Board pearl: The incretin effect accounts for 50-70% of postprandial insulin secretion — this is impaired in type 2 diabetes and exploited by GLP-1 agonists and DPP-4 inhibitors.

Insulin Clearance and Degradation
⭐
First-pass hepatic extraction removes ~50% of portal insulin, resulting in portal:peripheral insulin ratio of 3:1.
⭐
Insulin-degrading enzyme (IDE) in liver and kidney cleaves insulin at multiple sites; mutations cause hyperinsulinemia.
⭐
Receptor-mediated endocytosis leads to lysosomal degradation of insulin-receptor complexes.
⭐
Insulin half-life is 4-6 minutes; C-peptide half-life is 20-30 minutes due to minimal hepatic extraction.
⭐
Board pearl: Insulinomas cause endogenous hyperinsulinemia with elevated C-peptide, while exogenous insulin administration suppresses C-peptide through β-cell rest.

Pathophysiology of Type 1 Diabetes
✅
Autoimmune destruction of β cells mediated by T cells recognizing islet autoantigens (insulin, GAD65, IA-2, ZnT8).
✅
HLA-DR3/DR4 heterozygotes have highest genetic risk; environmental triggers (viruses, diet) initiate autoimmunity in susceptible individuals.
✅
Progressive β-cell loss → falling insulin production → hyperglycemia when ~80-90% of β cells are destroyed.
✅
Absolute insulin deficiency → unrestrained lipolysis and ketogenesis → diabetic ketoacidosis.
✅
Board pearl: Type 1 diabetes can present at any age — adult-onset requires positive autoantibodies (especially GAD) to distinguish from type 2.

Pathophysiology of Type 2 Diabetes
🧠
Begins with insulin resistance in muscle, adipose, and liver, initially compensated by β-cell hypersecretion.
🧠
Progressive β-cell dysfunction from glucolipotoxicity, ER stress, oxidative stress, and amyloid deposition → relative insulin deficiency.
🧠
Hepatic insulin resistance → unrestrained gluconeogenesis → fasting hyperglycemia.
🧠
Adipose insulin resistance → elevated free fatty acids → worsening insulin resistance in muscle and liver.
🧠
Board pearl: Type 2 diabetes requires both insulin resistance AND β-cell dysfunction — obesity alone causes insulin resistance but not diabetes if β-cells can compensate.

Insulin Therapy Principles
⚡
Exogenous insulin cannot replicate physiologic portal delivery or glucose-responsive secretion.
⚡
Rapid-acting analogues (lispro, aspart, glulisine) have modified B-chain sequences preventing hexamer formation → faster absorption.
⚡
Long-acting analogues (glargine, detemir, degludec) have modifications promoting slow, steady absorption → basal coverage.
⚡
NPH insulin forms crystals with protamine and zinc → intermediate duration with peak at 4-8 hours.
⚡
Board pearl: Insulin stacking (giving rapid-acting doses too close together) causes delayed hypoglycemia — the most common error in intensive management.

Clinical Assessment of β-Cell Function
📌
Fasting insulin and C-peptide levels reflect basal secretion; HOMA-IR = (fasting glucose × fasting insulin)/405 estimates insulin resistance.
📌
Oral glucose tolerance test: normal response shows insulin peak at 30-60 minutes; delayed or blunted peak indicates β-cell dysfunction.
📌
Mixed meal test provides physiologic stimulus incorporating incretins; more accurate than OGTT for β-cell function.
📌
Arginine stimulation test assesses maximum secretory capacity independent of glucose.
📌
Board pearl: In early type 2 diabetes, postprandial glucose rises before fasting glucose due to loss of first-phase insulin secretion.

Board Question Stem Patterns
📣
Obese patient with acanthosis nigricans and normal glucose → insulin resistance without diabetes.
📣
Lean patient with diabetes and low C-peptide → type 1 diabetes or late-stage type 2 with β-cell failure.
📣
Hypoglycemia with high insulin and high C-peptide → insulinoma or sulfonylurea use.
📣
Hypoglycemia with high insulin and low C-peptide → exogenous insulin administration.
📣
Diabetes with mild hyperglycemia and strong family history in non-obese patient → consider MODY.
📣
Neonate with severe hypoglycemia despite feeding → congenital hyperinsulinism from KATP channel mutation.
📣
Severe insulin resistance with normal or high insulin levels → consider receptor mutations or antibodies.

One-Line Recap
🔸
Insulin synthesis involves glucose-stimulated transcription → ER processing of preproinsulin → proinsulin cleavage in granules, while secretion requires glucose metabolism → KATP channel closure → Ca²⁺ influx → exocytosis, triggering receptor autophosphorylation → IRS recruitment → PI3K-AKT metabolic signaling and MAPK mitogenic signaling, with tissue-specific actions on glucose uptake, glycogen synthesis, lipogenesis, and gluconeogenesis that are impaired in diabetes through β-cell dysfunction and molecular mechanisms of insulin resistance.

bottom of page