top of page

eduo
visual

Reproductive & Endocrine Systems
Congenital adrenal hyperplasia
Core Principle of Congenital Adrenal Hyperplasia
🧷
Congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders caused by enzyme deficiencies in cortisol synthesis.
🧷
When cortisol production is impaired, loss of negative feedback causes ACTH to rise, driving adrenal hyperplasia and shunting steroid precursors into alternate pathways.
🧷
The specific enzyme deficiency determines which precursors accumulate and which hormones are deficient, creating distinct clinical phenotypes.
🧷
Board fundamental: CAH always involves cortisol deficiency with compensatory ACTH elevation — the varying presentations depend on which other hormones are affected.

The Adrenal Steroidogenesis Pathway
📍
Cholesterol → pregnenolone → 17-hydroxypregnenolone → DHEA (via 17α-hydroxylase)
📍
Pregnenolone → progesterone → 17-hydroxyprogesterone → androstenedione (via 21-hydroxylase)
📍
Progesterone → 11-deoxycorticosterone → corticosterone → aldosterone (mineralocorticoid pathway)
📍
17-hydroxyprogesterone → 11-deoxycortisol → cortisol (glucocorticoid pathway)
📍
Each enzyme block causes accumulation of precursors above the block and deficiency of products below.
📍
Board pearl: Draw this pathway — enzyme location determines the pattern of hormone excess and deficiency.

21-Hydroxylase Deficiency: The Classic CAH
🔹
Accounts for 90-95% of all CAH cases — the default diagnosis when CAH is mentioned without specification.
🔹
Blocks conversion of 17-hydroxyprogesterone → 11-deoxycortisol AND progesterone → 11-deoxycorticosterone.
🔹
Results in cortisol deficiency, aldosterone deficiency (in severe cases), and androgen excess from precursor shunting.
🔹
Elevated 17-hydroxyprogesterone is the diagnostic marker — this is the screening test for newborns.
🔹
Board pearl: 21-hydroxylase deficiency = ↓cortisol, ↓aldosterone, ↑androgens, ↑17-hydroxyprogesterone.

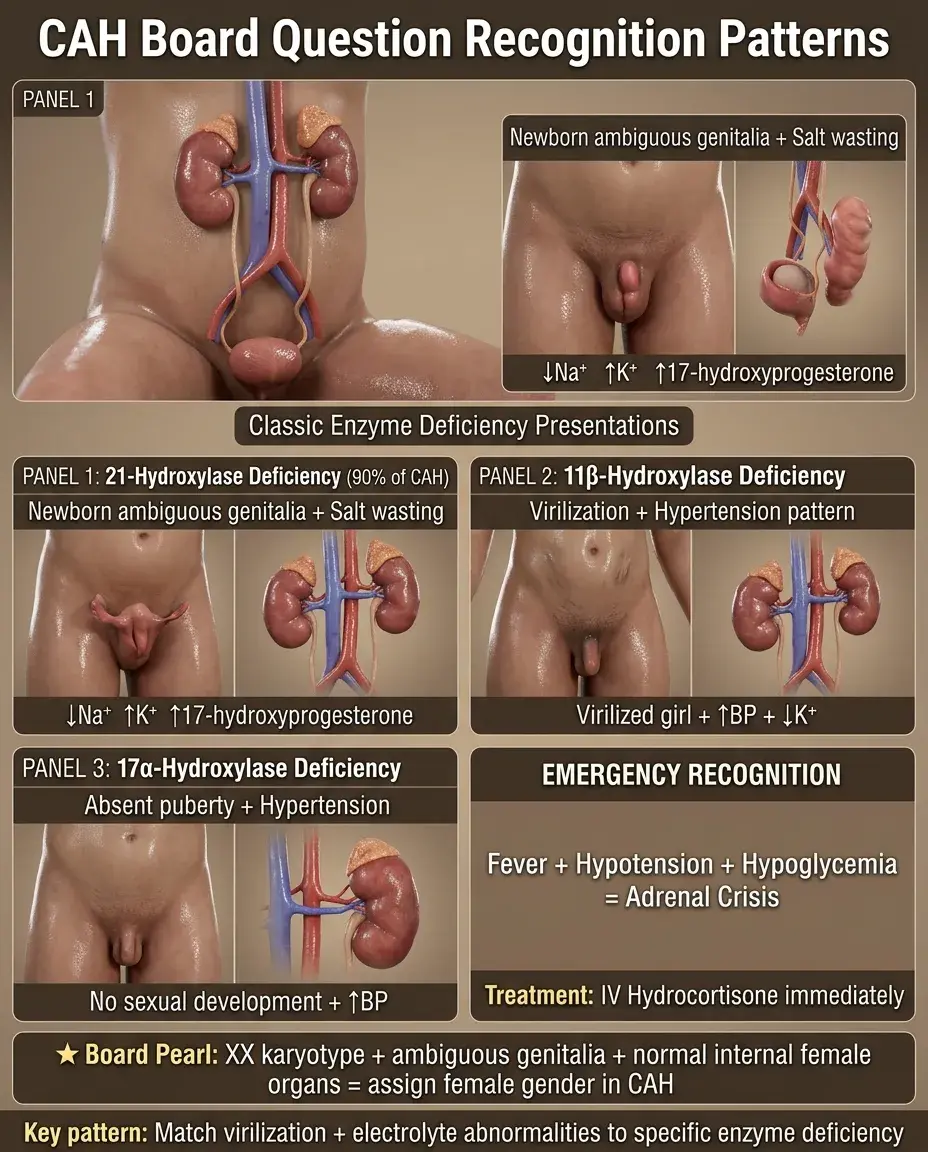
Clinical Presentations of 21-Hydroxylase Deficiency
⭐
Salt-wasting form (75%): severe enzyme deficiency → mineralocorticoid deficiency → hyponatremia, hyperkalemia, dehydration, shock in first 2-3 weeks of life.
⭐
Simple virilizing form (25%): partial enzyme activity preserves some aldosterone production → virilization without salt wasting.
⭐
XX infants: ambiguous genitalia at birth due to in utero androgen exposure (clitoromegaly, labial fusion, urogenital sinus).
⭐
XY infants: normal male genitalia at birth, but develop precocious puberty if untreated.
⭐
Board clue: Ambiguous genitalia in XX infant + salt wasting = classic 21-hydroxylase deficiency.

11β-Hydroxylase Deficiency: The Hypertensive CAH
✅
Second most common CAH (5-8%), blocking the final step of cortisol synthesis: 11-deoxycortisol → cortisol.
✅
Also blocks 11-deoxycorticosterone → corticosterone in the aldosterone pathway.
✅
11-deoxycorticosterone accumulates and has mineralocorticoid activity → hypertension and hypokalemia.
✅
Androgen excess still occurs due to precursor shunting → virilization.
✅
Board distinction: Virilization + hypertension + hypokalemia = 11β-hydroxylase deficiency (opposite electrolytes from 21-hydroxylase).

17α-Hydroxylase Deficiency: The Sexually Infantile CAH
🧠
Blocks both 17-hydroxylation (pregnenolone → 17-hydroxypregnenolone) AND 17,20-lyase activity (17-hydroxypregnenolone → DHEA).
🧠
Cannot produce androgens or cortisol, but mineralocorticoid pathway remains intact.
🧠
Precursors flood into mineralocorticoid synthesis → 11-deoxycorticosterone accumulation → hypertension and hypokalemia.
🧠
XX patients: normal female external genitalia but absent puberty (no sex hormones).
🧠
XY patients: female external genitalia (no testosterone), undescended testes, absent puberty.
🧠
Board pearl: Hypertension + absent puberty in either sex = 17α-hydroxylase deficiency.

Rare Forms of CAH
⚡
3β-Hydroxysteroid dehydrogenase deficiency: blocks conversion of all Δ5 to Δ4 steroids → severe cortisol and aldosterone deficiency with mild virilization from DHEA.
⚡
StAR protein deficiency (lipoid CAH): cannot transport cholesterol into mitochondria → complete absence of all steroid synthesis → severe salt wasting, XY sex reversal.
⚡
P450 oxidoreductase deficiency: affects multiple P450 enzymes → combined features of 17α-hydroxylase and 21-hydroxylase deficiencies.
⚡
Board approach: These are rare — focus on recognizing the three main forms.
Diagnosis of CAH
📌
Newborn screening: 17-hydroxyprogesterone elevation detects 21-hydroxylase deficiency.
📌
Confirmatory testing: measure specific precursors that accumulate above each enzyme block.
📌
ACTH stimulation test may unmask partial deficiencies by maximally stimulating the pathway.
📌
Genetic testing identifies specific mutations for family counseling.
📌
Prenatal diagnosis possible via amniocentesis or chorionic villus sampling in at-risk families.
📌
Board pearl: Elevated 17-hydroxyprogesterone = 21-hydroxylase; elevated 11-deoxycortisol = 11β-hydroxylase.

Treatment Principles of CAH
📣
Replace deficient hormones: hydrocortisone for cortisol deficiency, fludrocortisone for aldosterone deficiency.
📣
Suppress ACTH to prevent ongoing stimulation of androgen production — requires supraphysiologic glucocorticoid doses.
📣
Monitor growth velocity, bone age, and 17-hydroxyprogesterone levels to assess treatment adequacy.
📣
Stress-dose steroids required during illness or surgery to prevent adrenal crisis.
📣
Board concept: Treatment simultaneously replaces what's missing and suppresses what's excessive.

Management of Ambiguous Genitalia
🔸
XX infants with virilization: assign female gender, as they have ovaries and uterus with fertility potential.
🔸
Genital reconstruction surgery controversial — timing debated between early childhood vs. adolescence/adulthood.
🔸
Psychological support essential for patients and families navigating gender identity and sexual function.
🔸
XY infants typically have normal male genitalia and are raised male.
🔸
Board approach: Gender assignment based on karyotype and internal anatomy, not external appearance.

Late-Onset (Non-Classic) CAH
🧷
Partial 21-hydroxylase deficiency with residual enzyme activity → milder phenotype presenting later in life.
🧷
Presents in adolescence or adulthood with hyperandrogenism: hirsutism, acne, irregular menses, infertility.
🧷
No salt wasting or ambiguous genitalia — cortisol production usually adequate at baseline.
🧷
Diagnosis: elevated baseline or ACTH-stimulated 17-hydroxyprogesterone.
🧷
Board distinction: Young woman with PCOS-like symptoms + elevated 17-hydroxyprogesterone = late-onset CAH.

CAH and Adrenal Crisis
📍
All forms with cortisol deficiency risk life-threatening adrenal crisis during stress.
📍
Triggers: infection, surgery, trauma, or any physiologic stress exceeding the body's cortisol production capacity.
📍
Presentation: hypotension, shock, hypoglycemia, hyponatremia, hyperkalemia, vomiting, altered mental status.
📍
Management: immediate IV hydrocortisone (not dexamethasone — need mineralocorticoid activity), IV fluids, dextrose.
📍
Board emergency: CAH patient with fever and hypotension = adrenal crisis until proven otherwise.

Prenatal Treatment of CAH
🔹
Dexamethasone crosses the placenta and suppresses fetal adrenal androgen production.
🔹
Started before 9 weeks gestation in at-risk pregnancies to prevent virilization of affected female fetuses.
🔹
Must treat all pregnancies initially since fetal sex and affected status unknown early.
🔹
Discontinued if fetus is male or unaffected female after prenatal testing.
🔹
Controversial due to unnecessary exposure of unaffected fetuses and potential long-term effects.
🔹
Board concept: Prenatal dexamethasone prevents virilization but doesn't cure the underlying enzyme deficiency.

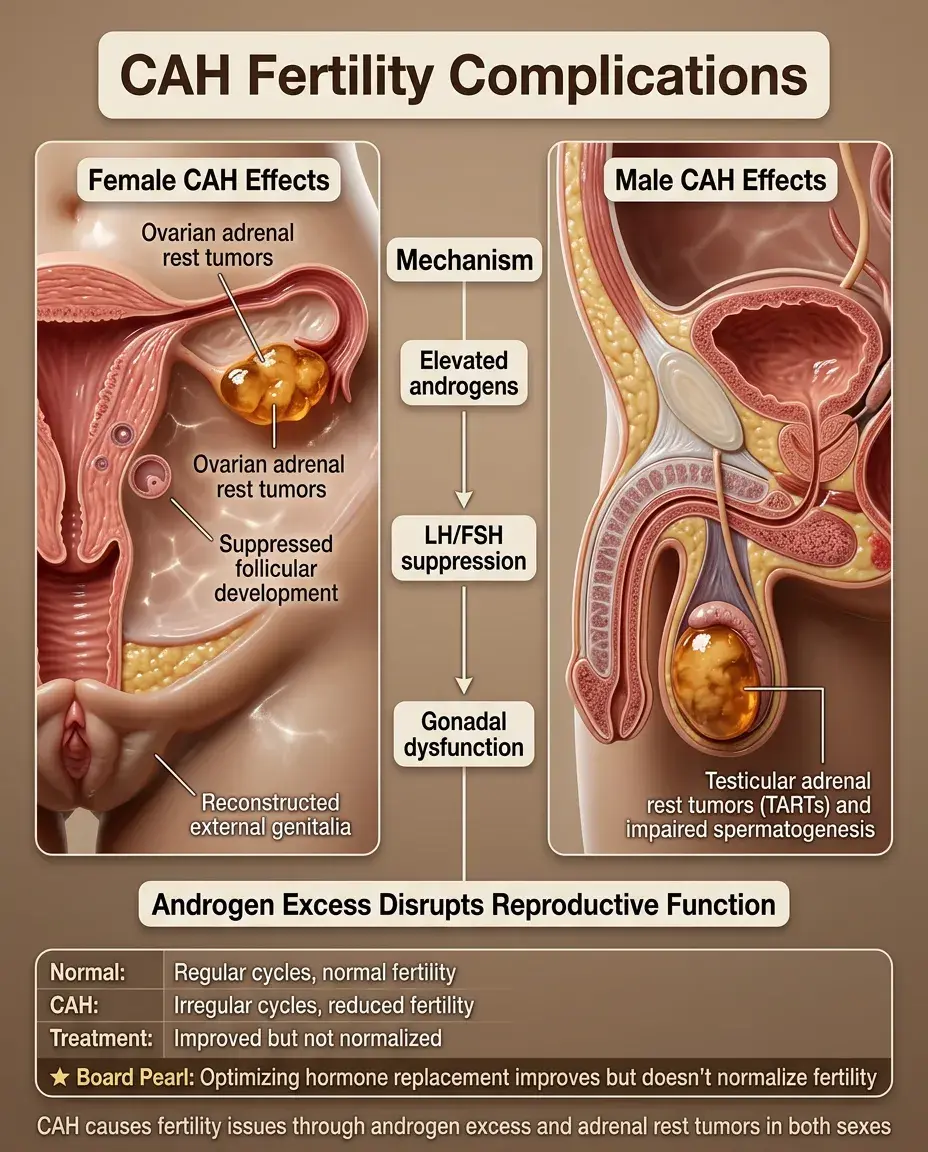
Fertility and CAH
⭐
Females with classic CAH have reduced fertility due to multiple factors.
⭐
Elevated androgens suppress gonadotropins and interfere with follicular development.
⭐
Adrenal rest tumors can develop in ovaries, impairing function.
⭐
Genital reconstruction may affect sexual function and vaginal delivery.
⭐
Males may develop testicular adrenal rest tumors (TARTs) causing infertility.
⭐
Board pearl: Optimizing hormone replacement improves but doesn't normalize fertility.
Molecular Genetics of CAH
✅
CYP21A2 gene mutations cause 21-hydroxylase deficiency — located in HLA complex on chromosome 6.
✅
Severity correlates with residual enzyme activity: null mutations → salt wasting; missense mutations → simple virilizing or non-classic.
✅
CYP11B1 mutations cause 11β-hydroxylase deficiency.
✅
CYP17A1 mutations cause 17α-hydroxylase deficiency.
✅
Genetic testing enables prenatal diagnosis and carrier screening in affected families.
✅
Board relevance: Autosomal recessive inheritance = 25% recurrence risk for carrier parents.

CAH Differential Diagnosis
🧠
Virilized female infant: CAH (most common), maternal androgen exposure, aromatase deficiency, androgen-secreting tumor.
🧠
Salt-wasting crisis: CAH, congenital adrenal hypoplasia, aldosterone synthase deficiency.
🧠
Ambiguous genitalia: CAH, androgen insensitivity syndrome, 5α-reductase deficiency, gonadal dysgenesis.
🧠
Precocious puberty: CAH, androgen-secreting tumors, McCune-Albright syndrome, familial male precocious puberty.
🧠
Board approach: Measure 17-hydroxyprogesterone first — elevated in >90% of CAH cases.

Complications and Monitoring
⚡
Short stature from premature epiphyseal fusion if undertreated (excess androgens) or overtreated (excess glucocorticoids).
⚡
Obesity and metabolic syndrome from chronic glucocorticoid therapy.
⚡
Osteoporosis risk with long-term supraphysiologic glucocorticoid doses.
⚡
Adrenal rest tumors in gonads — screen with ultrasound.
⚡
Psychological issues related to genital ambiguity, gender identity, and chronic disease.
⚡
Board monitoring: Growth velocity, bone age, and hormone levels guide therapy adjustments.

Board High-Yield CAH Patterns
📌
Salt wasting + virilization = 21-hydroxylase deficiency
📌
Hypertension + virilization = 11β-hydroxylase deficiency
📌
Hypertension + sexual infantilism = 17α-hydroxylase deficiency
📌
Elevated 17-hydroxyprogesterone = 21-hydroxylase deficiency
📌
Elevated 11-deoxycortisol = 11β-hydroxylase deficiency
📌
ACTH stimulation differentiates late-onset CAH from PCOS
📌
Memory framework: 21 loses salt, 11 gains pressure, 17 loses sex.

Board Question Stem Patterns
📣
Newborn with ambiguous genitalia + hyponatremia + hyperkalemia → 21-hydroxylase deficiency
📣
Virilized girl + hypertension + hypokalemia → 11β-hydroxylase deficiency
📣
Teenager with absent puberty + hypertension → 17α-hydroxylase deficiency
📣
Woman with hirsutism + elevated 17-hydroxyprogesterone after ACTH → late-onset 21-hydroxylase deficiency
📣
CAH patient with fever + hypotension + hypoglycemia → adrenal crisis requiring IV hydrocortisone
📣
Precocious puberty + advanced bone age + elevated 17-hydroxyprogesterone → untreated CAH
📣
XX karyotype + ambiguous genitalia + normal internal female organs → assign female gender
One-Line Recap
🔸
Congenital adrenal hyperplasia results from enzyme deficiencies in cortisol synthesis causing ACTH-driven adrenal hyperplasia with precursor shunting, presenting as three main patterns: 21-hydroxylase deficiency (salt wasting + virilization), 11β-hydroxylase deficiency (hypertension + virilization), and 17α-hydroxylase deficiency (hypertension + sexual infantilism), diagnosed by elevated precursor hormones and treated with hormone replacement to restore deficiencies and suppress androgen excess.

bottom of page